
CPC #4: Answer
The patient's electrocardiogram shows left ventricular hypertrophy based on the R wave amplitude in lead V6 of the EKG. Left ventricular hypertrophy occurs as a response to the pressure overload secondary to the coarctation. There is a known association of bicommissural aortic valve with aortic aneurysm; bicommissural aortic value, which occurs in 3~% of the population, is associated with a 9 to 18 fold increased risk of aortic aneurysm. Indeed, this patient does demonstrate a 4 cm diameter ascending aortic diameter (2.4 cm is normal diameter).
At autopsy, the patient demonstrated the classic finding of aortic dissection, with a tear in the posterior aorta leading to blood dissection into a false lumen, which extended retrograde back toward the heart. The aneurysm ruptured into the pericardium, yielding 600 ml of clotted blood (hemopericardium) that created tamponade clinically. The dissection also surrounded the aortic arch, and obstructed the right coronary artery, leading to terminal myocardial infarction. Autopsy confirmed the finding of left ventricular hypertrophy, as the heart weighed 400 grams (normal range 225 to 350 grams) as well as the presence of a bicommissural aortic value. Histologic sections of the aorta demonstrated cystic medial necrosis (disruption of the elastic lamina and deposition of mucopolysaccharide) similar to that which would be seen in a patient with Marfan's syndrome. However, these findings are not specific and may be seen in any cause of aortic dissection (See Figures Below).
Sudden death is rare in young people, though it has devastating impact. When violent causes (such as suicide, homicide, and accidents) and neoplasms are excluded, the main causes of sudden death in young people are pulmonary, neurologic and cardiac. Pulmonary causes include asthma, pneumonia, airway obstruction, and pulmonary hemorrhage secondary to adult respiratory distress syndrome. Neurologic causes include subarachnoid hemorrhage from rupture of aneurysms and arteriovenous malformations, parenchymal hemorrhage, seizures, and Arnold-Chiari malformations in which the cerebellum is displaced inferiorly and obstructs the cerebral-spinal fluid.
A long list of Cardiac causes of sudden death exists in young people. Arrythmogenic right ventricular dysplasia is more common in northern Italy. It is associated with replacement of myocardial cells in the right ventricular wall with fibrous and fatty tissue, resulting in a thin, dilated right ventricle which is associated with right ventricular tachyarrhythmia and sudden death. MRI can be very useful in establishing this diagnosis when it shows replacement of the myocardium with fat. Hypertrophic cardiomyopathy is associated with family history of affected first-degree relatives in 60% of cases. The remaining 40% likely represent new mutations. The thickened cardiac muscle obstructs of the cardiac outflow tract, and patients may present initially with syncope. However, most have no prodrome before sudden death. Dilated cardiomyopathy is familial in approximately 10% of cases and may represent late stage, resolving myocarditis. Myocarditis accounts for approximately 30% of sudden deaths from cardiac causes and is possibly associated with infection with Group B coxsackie viruses. Recent influenza-like illness is common. Coronary artery abnormalities that cause sudden death can be either congenital or acquired. The more frequent congenital coronary artery anomalies involve either an anomalous course or origin of either the right or left coronary artery. With anomalous course, there may be exertional ischemia leading to ventricular tachycardia, and fibrillation. Acquired coronary artery aneurysms most commonly occur in children secondary to Kawasaki's disease, which is a vasculitis of the coronary arteries that occurs most frequently in Asian patients. The congenital long-QT syndrome occurs with or without neurosensory deafness and is familial in 60% of cases. Ventricular tachycardia occurs in 80% of untreated patients. Aortic dissection is classically associated with Marfan's syndrome.
A variety of terms are frequently misused in relation to aortic dissections. It is important to understand the proper definitions of these terms. An aneurysm refers to a marked full thickness (all layers of the wall are present) enlargement. A dissection refers to a split in the intimal lining of the blood vessel with propagation of the blood within a false lumen. A tear is a post-traumatic event, most frequently occurring after the take-off of the left subclavian artery. A pseudoaneurysm is a contained area of blood accumulation and occurs with rupture of the wall. A dissecting aneurysm implies that there is dissection into an existing aneurysm, as defined above.
This patient never reached the operating room, but surgery remains the mainstay of treatment of aortic dissection. Prophylactically, dilatation of the ascending aorta to greater than 5.5 cm (in adults) is an indication to replace it with a Dacron tube. The threshold for replacement may be lower if the patient has a family history of aneurysms. Treatment of dissection depends upon whether the ascending aorta is involved. Involvement of the ascending aorta (Stanford Group A) is an indication for immediate surgery. Patients with dissections that propagate distally into the descending aorta (Stanford Group B) are treated surgically only if there are complications, such as rupture, persisting pain (indicative of impending rupture), and evidence of malperfusion of organs such as the kidney. Medical therapy is more efficacious in this latter setting.
This patient did not demonstrate
clinical evidence of Marfan's syndrome.
However, it is likely that she suffered
from a deficiency in one of the
proteins that help maintain the
integrity of elastin and its attachment
to cells, as fibrillin-1 (the defective
protein in Marfan's syndrome) does.
It is becoming apparent that the
association of aortic aneurysm and
biccommissural aortic valve is not
a consequence of hemodynamic perturbations
secondary to the bicuspid valve.
Rather, each of these is a primary
abnormality due to a single gene
defect. The identity of this gene
is a subject of ongoing research.
Cystic
Medial Necrosis of Aorta -Hematoxylin
and Eosin Stain

Alcian
Blue Stain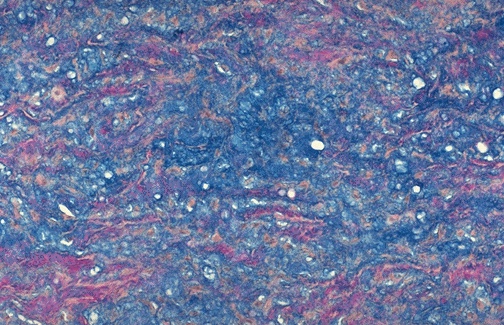 |