
Answer to CPC #1 (Tuesday, September 24, 2002)
This patient's presentation was marked by a variety of non-specific symptoms and signs, which makes arriving at a diagnosis difficult. However, from the clinical history, we can distill the presentation down to several core symptoms and signs. First, the patient suffers from a normocytic anemia and marked thrombocytopenia, in the face of a normal white cell blood count. Therefore, two of the three blood cell lines are diminished. The patient shows no signs of blood loss through the gastrointestinal tract. Second, the patient has elevated AST and LDH in the background of normal liver function tests, which raises the possibility of hemolysis since the former two enzymes are present in red blood cells. Third, the patient has normal coagulation parameters with normal fibrinogen, PT and aPTT, indicating that the patient does not have disseminated intravascular coagulopathy. Fourth, the patient suffers from acute renal failure. Fifth, the patient demonstrates signs of mental status changes, in the form of lethargy and malaise.
In this setting, it is sometimes
easier to approach the differential
diagnosis for several of the key
symptoms and signs of the patient.
One possibility is to approach the
patient's thrombocytopenia, and
come up with a differential diagnosis.
Possibilities here include decreased
production that could arise from
a viral infection, bone marrow infiltration
by malignancy or granulomatous inflammation,
or bone marrow failure in the setting
of myelodysplastic syndrome or acute
leukemia. More commonly, thrombocytopenia
arises from increased destruction,
such as occurs from ITP (which can
be idiopathic or drug associated),
infection (either overwhelming sepsis
or viral infection), malignancy,
or consumption. Platelet consumption
occurs by either disseminated intravascular
coagulation or a microangiopathic
hemolytic anemia. A bone marrow
exam, to look for increased or decreased
megakaryocytes, or a platelet transfusion,
to indicate shortened platelet survival,
is sometimes necessary to determine
the etiology of thrombocytopenia.
Thus, it is difficult to narrow
down this patient's differential
diagnosis from her thrombocytopenia.
Alternatively, we should approach this patient's differential diagnosis by examining the possible causes of her anemia. The differential diagnosis for anemia can be divided into hypoproliferative states and hyperproliferative states. Hypoproliferative anemias are generally characterized by a low reticulocyte count. The differential diagnosis includes deficiencies of the building blocks of red blood cells (Vitamin B-12, folate or iron), parvovirus infection (which specifically targets erythroid precursors), bone marrow failure or infiltration (such as with leukemia or granulomatous disease, respectively) or chronic inflammation which gives rise to the anemia of chronic disease. Hyperproliferative anemias are characterized by increased reticulocyte counts that cannot compensate for increased destruction or blood loss. The patient shows no evidence of blood loss through the gastrointestinal tract or elsewhere. Hyperproliferative anemias resulting from red blood cell destruction, or hemolysis, can be either extravascular or intravascular. Extravascular destruction occurs in the spleen, as antibodies coat red blood cells leading to phagocytosis in the red pulp. This type of hemolysis is associated with a positive Coombs test and microspherocytes on peripheral smears; the latter results from the pinching off of fragments of cell membrane by the spleen's sinusoidal macrophages. Intravascular causes of hemolysis are associated with increased LDH and AST (which are released from red blood cells) and decreased haptoglobin levels. Intravascular hemolysis is often associated with low platelet counts (consumptive thrombocytopenia). Intravascular hemolysis is characterized by schistocytes on peripheral smear.
Examination of the provided peripheral blood smear helps greatly to characterize this patient's anemia. The first finding of note is the markedly decreased platelet count. On a blood smear, each platelet seen in a high power field correlates with approximately 10,000 platelets in the peripheral circulation. Hence, since the normal platelet count is approximately 140,000, one should see approximately 14 platelets in a high power field, and we see far fewer than that in the provided peripheral smear images. We note that the few white blood cells seen in the smear appear normal, which correlates with the normal white count noted in the peripheral smear. The most striking finding in the blood smear, however, relates to the red blood cells. We see indirect evidence of reticulocytosis in the marked polychromasia of the red blood cells. While we see a few microspherocytes, the dominant finding is that of numerous schistocytes and helmet shaped cells. The presence of these numerous schistocytes establishes that this patient has a microangiopathic hemolytic anemia.
Microangiopathic hemolytic anemias occur from mechanical shearing of the red cells, and consumption of platelets, and the differential for this syndrome is limited. One of the more common causes is disseminated intravascular coagulation (DIC). In a patient with a skin rash, as the current patient had, meningococcal sepsis leading to DIC is a possibility. However, patients with DIC typically have abnormal coagulation factors (increased PT and aPTT, decreased fibrinogen), which this patient did not have. Patients with sepsis also typically have fever and hypotension. Another possibility is the HELLP (hemolysis, elevated liver enzymes, low platelets) syndrome, which may develop in pregnant patients and is associated with marked liver function abnormalities. Neither of these was true of the current patient. Another possibility is the "waring blender syndrome", in which hemolysis is associated with heart valve abnormalities. The absence of a cardiac murmur or prior valve replacement argues strongly against this possibility. Occasionally, patients with vasculitis, lupus or antiphospholipid syndrome present with microangiopathic hemolytic anemia; however, this patient did not demonstrate any signs of these disorders. Yet another possibility is malignant hypertension, though this is eliminated by the normal blood pressure.
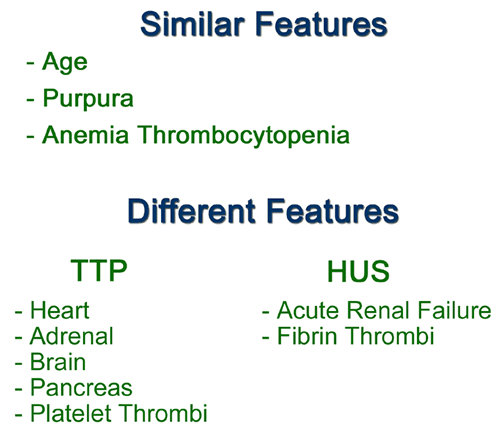
The final category of microangiopathic hemolytic anemia includes the distinct but related, thrombotic syndromes, thrombotic thrombocytopenic purpura (TTP) and the hemolytic-uremic syndrome (HUS). The syndromes are characterized by thrombocytopenia, hemolytic anemia, fever, renal abnormalities, and neurologic abnormalities. HUS is characterized by greater renal dysfunction, less severe thrombocytopenia, less elevation of LDH, and fewer schistocytes than TTP, while TTP is characterized by predominant neurologic abnormalities, but these disorders may have significant clinical overlap. While most cases are idiopathic, these disorders have several associations. TTP can be familial, associated with HIV infection, or associated with bone marrow transplants or medications (such as cyclosporine and ticlopidine). There is an association of HUS with the Shiga toxin produced by E. coli 0157:H7. The classic pentad associated with TTP is microangiopathic hemolytic anemia, thrombocytopenia, fever, mental status changes and renal dysfunction. This patient lacked significant fever, but it is important to know that approximately 50% of patients with TTP will not have all of the five signs of the pentad of TTP. Other subtle features of TTP include the fact that the white cell blood cell count is typically normal, liver and lung functions are typically normal, and the mental status involvement is not chronic, as patients may rapidly return to normal CNS function after being essentially comatose.
Given this patient's consolation of clinical findings, the most likely diagnosis is TTP. The treatment of TTP involves plasmapheresis, which has been thought to work by removing an abnormal factor which promotes coagulation from the blood and/or restoring an absent factor to it. This patient was treated for TTP with plasmapheresis, but despite therapy expired suddenly. An autopsy was performed.
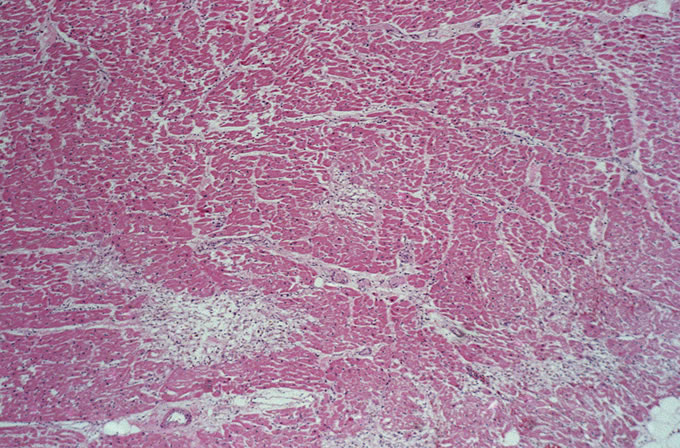
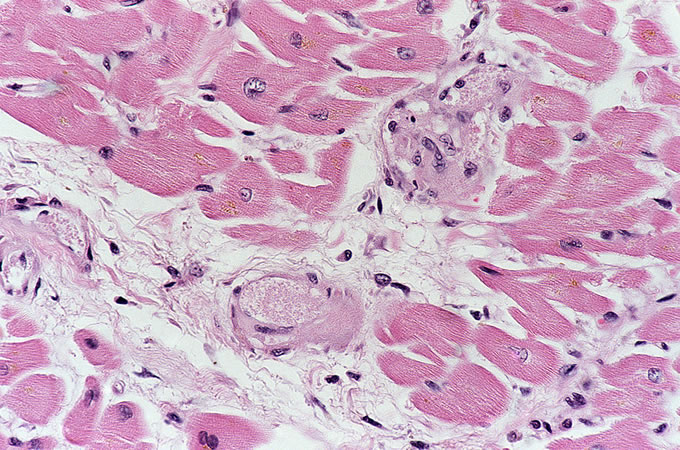
The autopsy findings confirmed the diagnosis of TTP. Examination of multiple organs revealed small arterioles with organizing platelet thrombi. These were most evident in the heart, where they were associated with small foci of myocyte loss with replacement fibrosis (Figure 1). Higher power examination of these thrombi revealed evidence of organization with endothelial cell proliferation, consistent with this patient's onset of symptoms approximately 8-9 days before demise (Figure 2).
Figure
1
Figure
2
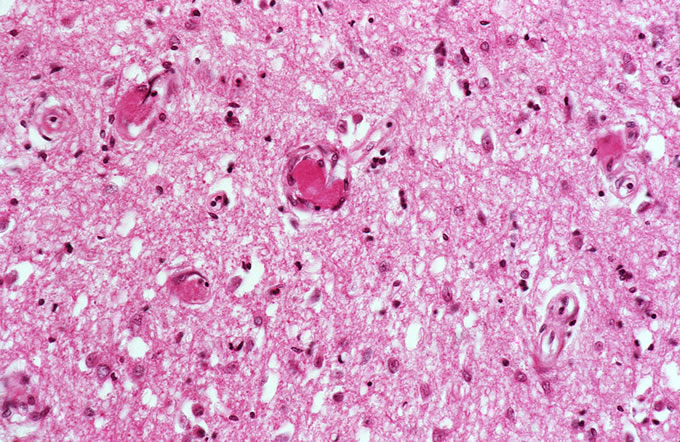
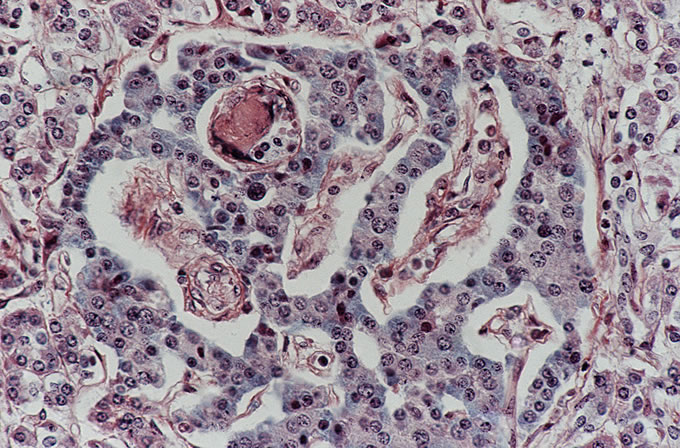
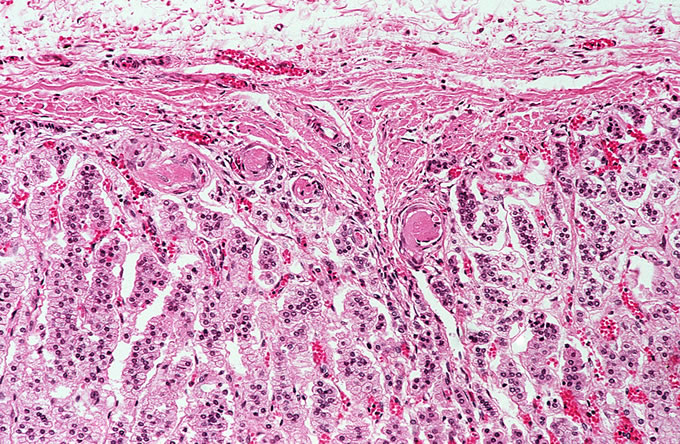
A recent autopsy series from this hospital has compared and contrasted the autopsy findings in a large series of patients with TTP and HUS. The study found numerous similar clinical features between the two types of patients, including similar age, presence of purpura, anemia and thrombocytopenia. However, several key differences, which reached statistical significance, were noted. As was seen in the current case, patients with TTP were far more likely to have thrombi affecting the heart. Thrombi in the brain (Figure 3) were more common in TTP, which correlates with the clinical presentation with mental status changes. The pancreas was more likely to be involved with TTP, and thrombi classically affect the small venules within islets of Langerhans (Figure 4, Figure 5). The adrenal was also more frequently involved in TTP, and the platelet thrombi characteristically affect the peripheral zona glomerulosa of the adrenal cortex (Figure 6). The platelet-rich composition of the thrombi in TTP can be confirmed by special stains. The thrombi are strongly immunoreactive for von Willebrand factor by immunohistochemistry (Figure 7), but do not label well with the PTAH stain, which is a good marker for fibrin (Figure 8). In contrast, the thrombi in HUS are more likely to be true fibrin thrombi, which label with the PTAH stain (Figure 9). Clinically, HUS was far more likely to be associated with acute renal failure than TTP (Figure 10).
Figure
3
Figure
4
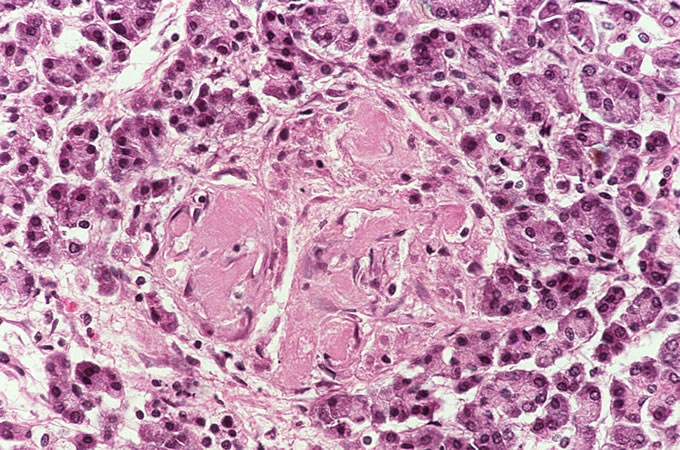
Figure
5
Figure
6
Figure
7
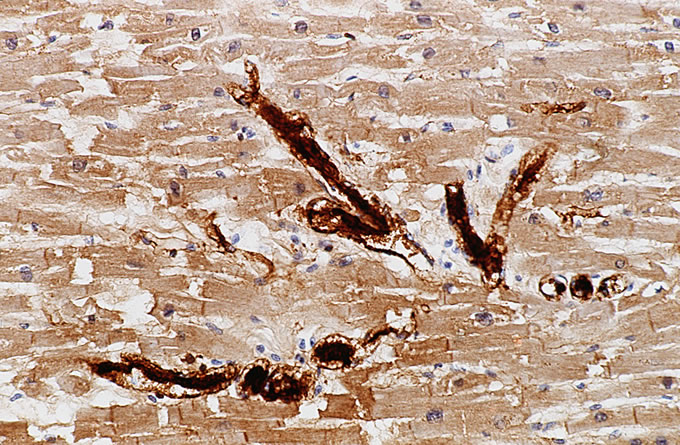
Figure
8
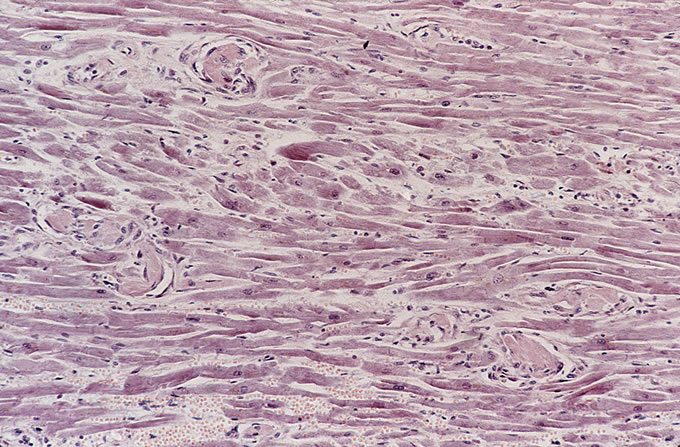
Figure
9
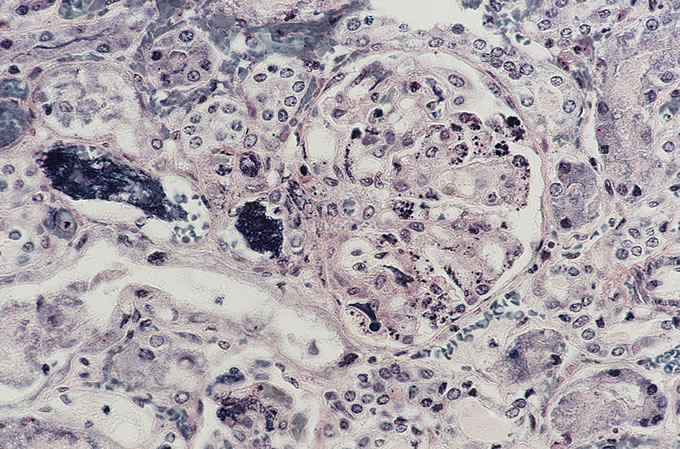
Figure
10
Recently, the pathogenesis of TTP has become more apparent. It had long been known that patients with TTP had unusually large von Willebrand factor multimers in their plasma, which were postulated to promote abnormal platelet aggregation and cause thrombi. Non-familial acquired TTP has been found to be associated with an acquired inhibitory antibody against the von Willebrand factor-cleaving metalloprotease, which normally cleaves such multimers. Familial forms of TTP are associated with a constitutional deficiency of this protease. In contrast, the HUS is not associated with these antibodies, and thus this single laboratory test may help us distinguish 2 syndromes that overlap clinically. It is hoped that this new knowledge may lead to more rational therapies for TTP in the future. One possibility is the direct administration of the inhibited protease (also known as ADAMTS 13).

|
References New
England Journal of
Medicine, 2002;
347:589-600. |